CASP is the Critical Assessment of Protein Structure Predictions, a biannual “competition” to determine which prediction algorithm generates the most accurate model. There are several categories in which models will be assessed, including accuracy, topology, and biological relevance. The SIBYLS beamline is participating to provide small-angle X-ray scattering (SAXS) data for––and judging for the first time––the “data-assisted” category. This CASP competition should lead to improvement in predicting protein-protein interfaces and complex structures.
Cryo-EM Reveals Structure of Human Telomerase
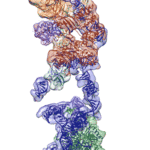
Regulation of the enzyme telomerase has been implicated in cancer, other human diseases, and aging, but progress towards therapeutic manipulation of telomerase has been hampered by the lack of high-resolution structural data. Now, researchers in senior faculty scientist Eva Nogales’ lab in Molecular Biophysics and Integrated Bioimaging (MBIB), in collaboration with UC Berkeley professor of Biochemistry, Biophysics and Structural Biology Kathleen Collins, have published a paper in Nature describing the 3-D molecular structure of the human telomerase enzyme.
Crystallization Screen Created by Berkeley Lab Biosciences Scientists Reaches the Market

 X-ray crystallography has been the most successful technique used to solve macromolecular structures, contributing several thousand new entries to the Protein Data Bank (PDB) every year. The protein crystal is the critical starting point for X-ray data collection, and consequently, its properties are correlated with the quality of the data and the level of detail that can be extracted for a macromolecular structure. However, proteins require solutions of specific composition to form crystals for structure determination studies. These specifications are usually determined from exposing the protein to several different solutions in a crystallization screen.
X-ray crystallography has been the most successful technique used to solve macromolecular structures, contributing several thousand new entries to the Protein Data Bank (PDB) every year. The protein crystal is the critical starting point for X-ray data collection, and consequently, its properties are correlated with the quality of the data and the level of detail that can be extracted for a macromolecular structure. However, proteins require solutions of specific composition to form crystals for structure determination studies. These specifications are usually determined from exposing the protein to several different solutions in a crystallization screen.
A team of researchers in the Molecular Biophysics and Integrated Bioimaging (MBIB) Division led by Paul Adams and Jose Henrique Pereira have developed a new crystal screen, the Berkeley Screen, with 96 conditions proven to be highly effective at producing crystals for structural determination. The Berkeley Screen is now available to the wider crystallography community commercially.
NIH Awards $6.5 Million for Augmenting Structural Biology Research Experience

The National Institutes of Health (NIH) has awarded $6.5 million to Berkeley Lab to integrate existing synchrotron structural biology resources to better serve researchers. The grant will establish a center based at the Lab’s Advanced Light Source (ALS) called ALS-ENABLE that will guide users through the most appropriate routes for answering their specific biological questions.
A Near-Atomic Resolution Map of a Key DNA Protein Complex

Molecular Biophysics and Integrated Bioimaging (MBIB) Division scientists led by Eva Nogales have resolved the 3-D structure of a critical human cellular protein complex involved in DNA transcription and repair at an unprecedented level of resolution. The complex, called transcription factor IIH (TFIIH), unzips the DNA double helix so that genes can be accessed and read. Malfunctions of the complex are associated with premature aging, cancer propensity, and a variety of other defects. One challenge with solving the structure of TFIIH has been that it exists in such minute amounts that it is difficult to produce and purify in large quantities. Moreover, once obtained, it may not form crystals suitable for X-ray diffraction. The researchers used cryo-electron microscopy (cryo-EM), a technique in which purified samples are flash-frozen at ultra cold temperatures, and which works even on very small quantities. “The fact that we resolved this protein structure from human cells makes this even more relevant to disease research,” said Nogales. Basil Greber, a postdoctoral fellow in Nogales’s lab, was first author on the study published in the journal Nature. Computational research scientist Pavel Afonine and MBIB Division Director Paul Adams also contributed to the project. Read more from the Berkeley Lab News Center.
Was this page useful?
Send