Skip to main content
Staff Portal
About Biosciences
Leadership
Biosciences Area Strategy
Biosciences Program Development Group
Biosciences Area History
Contact Information
Our Science
Biosciences Research
Strategic Initiatives
Biological Systems and Engineering
Environmental Genomics and Systems Biology
Molecular Biophysics and Integrated Bioimaging
DOE Joint Genome Institute
Media and Events
News
Behind the Breakthroughs
Events Calendar
Seminar Series
Work With Us
Work With Us
Organizational Stewardship
Staff Portal
About Biosciences
Leadership
Biosciences Area Strategy
Biosciences Program Development Group
Biosciences Area History
Contact Information
Our Science
Biosciences Research
Strategic Initiatives
Biological Systems and Engineering
Environmental Genomics and Systems Biology
Molecular Biophysics and Integrated Bioimaging
DOE Joint Genome Institute
Media and Events
News
Behind the Breakthroughs
Events Calendar
Seminar Series
Work With Us
Work With Us
Organizational Stewardship
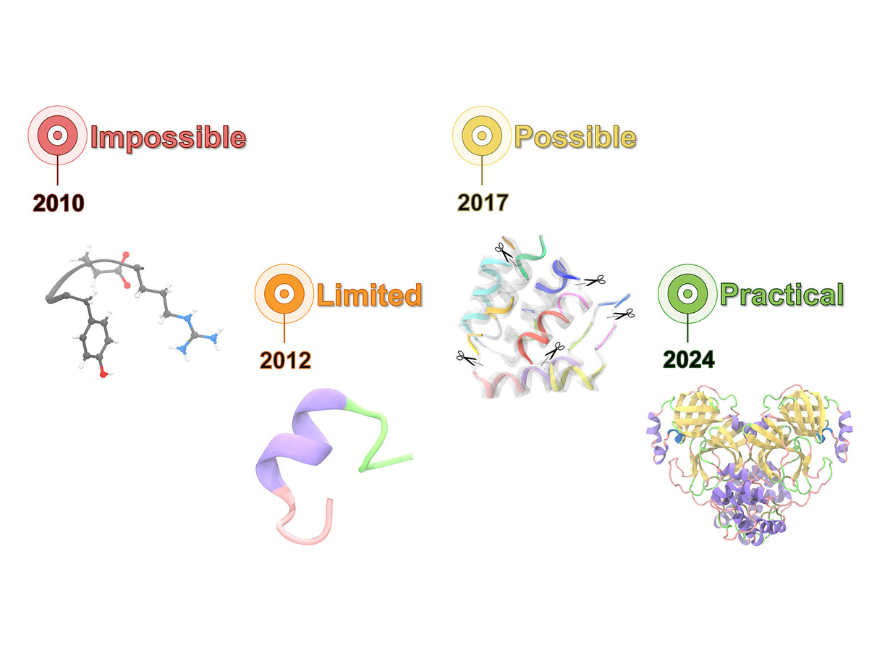
Article
Molecular Biophysics and Integrated Bioimaging
Article
Molecular Biophysics and Integrated Bioimaging
Article
Molecular Biophysics and Integrated Bioimaging
Article
Molecular Biophysics and Integrated Bioimaging
Article
Biosciences Area
Article
Molecular Biophysics and Integrated Bioimaging
Article
Molecular Biophysics and Integrated Bioimaging
Article
Article
Article
Article
Article
Posts pagination
1
2
>
Was this page useful?
Send
About Us
Leadership
Biosciences Research
Work With Us
Contact
Resources
A-Z Index
Phonebook
Acronyms
Integrated Safety Management