
The U.S. National Institutes of Health (NIH) and other funding agencies around the world have invested vast resources to ultimately sequence the complete genomes of millions of individuals with various common and rare diseases in order to identify underlying genetic causes. However, this work thus far has identified the disease-causing genetic changes in only a small number of patients. Because protein-coding genes comprise less than 3% of the human genome, geneticists increasingly suspect that mutations to non-coding DNA may be the culprit in a lot of these cases.
The effects of mutations in protein-coding genes are predictable because we understand the genetic code, but it’s more difficult to assess the functional consequences of mutations in non-coding sequence. Scientists know that non-coding sequence contains regulatory switches, called enhancers, that control when and where genes are turned on or off in an organism. A mutation within one of these enhancer sequences may cause the activity of the gene it controls to be too high, too low, or to be misdirected to a cell type or tissue where it may have detrimental effects on the organism.
While it is understood, in principle, that a sequence change in an enhancer can cause disease, identifying such a mutation remains a major hurdle. Sequencing the genomes of many individuals has revealed that each person’s genome contains dozens to hundreds of new mutations compared to their parents’ genomes. Yet, the majority of these changes don’t disrupt normal gene regulation. The challenge is to identify the small subset of mutations that change the sequence of specific enhancers in a deleterious way.
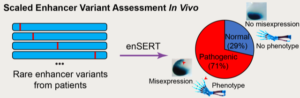
New work from the Mammalian Functional Genomics Laboratory in Biosciences’ Environmental Genomics and Systems Biology (EGSB) Division addresses this critical challenge. The group has developed a higher-throughput transgenic mouse assay to evaluate the disease-causing potential of human variants in enhancers that turn on gene expression during development. The new approach leverages the CRISPR-Cas9 genome editing technology to create transgenic mice that carry an enhancer-reporter construct at a specific “safe harbor” location in the mouse genome. A color-generating chemical reaction (the “reporter”) creates a blue stain in all cells in which the enhancer is active.

From left: Diane Dickel, Evgeny Kvon, Len Pennacchio, and Axel Visel of the Mammalian Functional Genomics Lab.
“Before, people would inject that enhancer-reporter into the mouse zygotes and it would randomly integrate in the genome,” said Evgeny Kvon, a project scientist in the EGSB Division and first author on the paper published in Cell. “And because of that random integration, the results were less reproducible because, depending on the integration site, there were so-called position effects that would affect the activity of that reporter.
“The major conceptual advance in our method is that because the transgenes are integrated in the same location in the genome there are no position effects, so we need fewer mice to get reproducible results,” he continued. What’s more, the researchers were fortunate to find a location in the mouse genome where the integration frequency is four times higher than with the old method, Kvon said. “So not only do we get more reproducibility but it’s also high efficiency. And that reduces the cost of performing this experiment several fold.”
![]()
To demonstrate proof of principle, the researchers used the new method—which they dubbed enSERT (enhancer inSERTion)—to examine nearly a thousand variants of one of the most well-characterized human enhancers that is associated with polydactyly (extra fingers or toes). The enhancer, called ZRS (Zone of polarizing activity Regulatory Sequence), regulates the expression of the sonic hedgehog gene (Shh), which produces a powerful signaling molecule required for the correct patterning of many body elements, including limbs and digits.
The ZRS enhancer is widely conserved across vertebrate species, including humans, mice, fish and even snakes. Mutations in this enhancer are known to cause abnormal patterning of the limbs in humans, as well as other vertebrates. The most common type of malformation caused by these mutations is called “preaxial polydactyly,” i.e. the formation of extra digits near the thumb or big toe. For example, mutations in the ZRS enhancer have been observed in polydactyl Hemingway cats, so-named for the American author and ailurophile Ernest Hemingway, who kept a large colony of the mitten-pawed felines at his Key West, Florida, home.

Polydactyl cat at Hemingway House in Key West, Florida.
Using the new assay, the researchers examined all human ZRS enhancer mutations that had previously been reported by other groups as potential causes of polydactyly, whether the proposed malformation-causing mechanism could be confirmed experimentally or not. They also evaluated additional sequence changes that clinician-scientists collaborating on this study had newly identified in patients with polydactyly. Overall, in about 70 percent of cases the researchers were able to confirm that the enhancer activity was changed by the mutations. Perhaps surprisingly, they found no evidence for changes in activity for the remaining 30 percent of cases, suggesting that a subset of mutations that were proposed to cause polydactyly in previous studies are not the true cause of the condition in these patients.
“These results suggest that extreme care should be taken when interpreting human mutations without experimental testing, because it is possible that one could be misled to think that they are causative when they could potentially just be one of the many rare benign mutations in the human population,” cautioned Diane Dickel who, along with Mammalian Functional Genomics Lab co-PIs Len Pennacchio and Axel Visel, was a corresponding author on the Cell paper.
The work supports the Berkeley Lab Biosciences Area’s health strategy, which aims to increase understanding of human genome function. “We expect that this method will be very powerful for systematically testing enhancer mutations that are being found by the large number of patient sequencing studies,” Dickel said. The group has some pilot projects in the lab that show the utility of the approach to experimentally validate non-coding mutations associated with a variety of conditions that significantly impact human health, such as developmental delay, autism, or heart disease.
Institutional collaborations on the study included the National Center for Biotechnology Information (NCBI) of the National Library of Medicine branch of the National Institutes of Health (NIH), Washington University School of Medicine in St. Louis, and several teaching hospitals (Centre hospitalier universitaire; CHU) in France: CHU Lille, CHU Poitiers, and CHU Dijon Bourgogne.

