Berkeley Lab geneticist Len Pennacchio and his team helped a group of Israeli clinical researchers solve the mystery of a rare inherited disease that causes extreme, sometimes fatal, chronic diarrhea in children. The nearly decade-long investigation not only led to the discovery of a novel protein-coding gene that is critical for intestinal function, but also expanded our understanding of regulatory sequences in the human genome. The results were recently published in Nature.
Intractable Diarrhea of Infancy Syndrome (IDIS) was first described in 1968 and affects just seven Jewish family lineages, but it may share disease-causing mechanisms with other gastrointestinal illnesses (such as irritable bowel or Crohn’s disease) that affect millions of people. At the time the clinical team’s lead researcher, Yair Anikster, began studying the disease in 2000, the only established information was that IDIS is a single-gene mediated recessive trait. Anikster and a diverse group of colleagues performed gene sequencing on samples from eight individuals who represented each of the seven affected families. They found that all of them harbored deletions in both copies of a previously unstudied noncoding region on chromosome 16.

From top: Len Pennacchio, Diane Dickel, and Axel Visel
Realizing they needed expert insight to establish a causal link between these deletions and IDIS, the Israeli group reached out to Pennacchio, whose Mammalian Functional Genomics Lab has been examining regulatory regions since wrapping up work on the landmark Human Genome Project. First Pennacchio, along with his co-PIs Diane Dickel and Axel Visel, and members of their team, used their mouse model platform to confirm that the chromosome 16 region is indeed a regulatory sequence involved in the development of the gastrointestinal system. When they engineered a lineage of mice with chromosomal deletions equivalent to those of Anikster’s human patients, the infant animals displayed diarrhea. These findings provided evidence that the noncoding sequence, which they termed the intestine-critical region (ICR), is the cause of the disease.
Then, after years of painstaking experimentation, the authors were able to determine that the ICR controls expression of a previously unknown nearby gene, now called Percc1. Looking back at their human sequence data, the authors saw that ICR deletions carried in IDIS families prevent any Percc1 protein from being made. To prove that a lack of Percc1 gene expression is the sole cause of IDIS, they bred a new lineage of mice that had no genetic abnormalities other than a deleted Percc1 gene. Immediately after birth, the mutant mice experienced severe, chronic diarrhea that perfectly mirrored the disease progression of humans with IDIS.
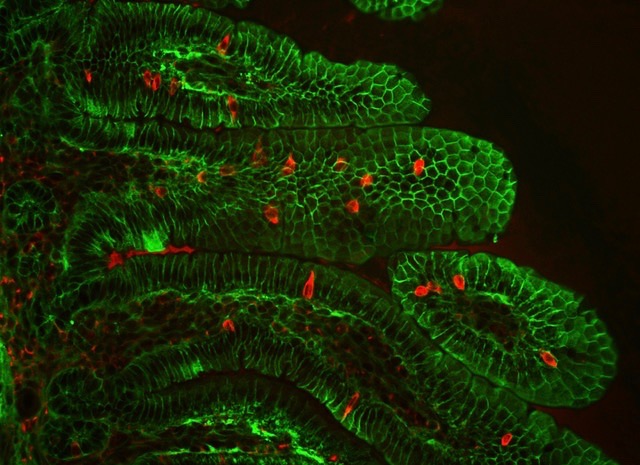
Detection of Percc1 expressing cells (red) amongst the epithelial cells (green) of the intestinal villi in a mouse. (Credit: Marco Osterwalder/Berkeley Lab)
Finally, using their mouse model and human stem cells, the scientists showed that the Percc1 protein is expressed in–and necessary to the proper functioning of–a very small group of hormone-producing gastrointestinal cells. Through chemical signaling, these cells appear to mediate many processes, including intestinal motility, glucose uptake, and triggering the sense of satiety.
Read more in the Berkeley Lab News Center.

