A team of researchers develops software to solve detailed structures automatically.
Within each cell of the human body, thousands of molecular machines are at work. They transport nutrients and biochemicals into and out of our cells, build other tiny machines, and even move our cells around. To understand how these molecular machines work, scientists create three-dimensional pictures using electron cryomicroscopy (cryo-EM), catching these machines in different shapes that give insight into their function. Now researchers at Berkeley Lab and their international collaborators who write and distribute the Phenix software suite have developed a new set of computational tools for automated structure determination from cryo-EM data.

Paul Adams, Pavel Afonine, and Oleg Sobolev are the Berkeley Lab computational researchers who developed the Phenix cryo-EM tool set software.
“Until recently, most of the 3D pictures of molecular machines have been obtained using a technique called X-ray crystallography,” said Paul Adams, Director of the Molecular Biophysics and Integrated Bioimaging (MBIB) Division. Adams leads the group of scientists at Berkeley Lab as well as the New Mexico Consortium, Los Alamos National Laboratory (LANL), Duke University, and Cambridge University, who develop Phenix. X-ray crystallographers around the world have been using Phenix to help obtain and interpret 3D pictures for nearly two decades.
In the past few years, exciting improvements in cryo-EM have allowed it to become as powerful for studying molecular machines as X-ray crystallography has been. Cryo-EM is a vital technology for meeting grand challenges in energy, environment, and health by developing new approaches for creating biofuels and advancing understanding of biological systems. “Thousands of researchers are now switching to this newer imaging method, but software for interpreting the 3D pictures from cryo-EM was missing,” said Adams.
The Phenix developers realized that their software could be modified to meet that need. “We knew we could adapt our programs by taking into account the differences between pictures obtained with X-ray crystallography and electron microscopy,” Adams said. One of the big differences is that the pictures from electron microscopy are usually less detailed than those from X-ray crystallography. “The software for [electron microscopy] has to be able to guess some details about the molecules without being able to see them clearly,” Adams continued.
In a paper that appeared last November in Nature Methods, the Phenix developers, led by Tom Terwilliger (pictured, left) of the New Mexico Consortium and LANL, presented their new software for identifying all the parts of a molecular machine, even creating a molecular model of the machine. A key feature of the software is that it is automatic. “It’s simple,” said Adams. “The researcher uploads a 3D picture of their molecule obtained with cryo-EM–along with the protein or nucleic acid sequence–and the software, which is now part of Phenix, finds all the parts of the molecule and how they are connected to each other.” Phenix then produces a picture of all the parts of the molecule that researchers can use to understand how their molecular machine works.
A key feature of the software is that it is automatic. “It’s simple,” said Adams. “The researcher uploads a 3D picture of their molecule obtained with cryo-EM–along with the protein or nucleic acid sequence–and the software, which is now part of Phenix, finds all the parts of the molecule and how they are connected to each other.” Phenix then produces a picture of all the parts of the molecule that researchers can use to understand how their molecular machine works.
This latest publication capped a series of related research articles describing the cryo-EM tools to aid in the structure solution process, from optimizing the initial map to validating the structure model obtained.
When building an atomic model from the cryo-EM map image, electron microscopists have to sharpen their maps with an optimal sharpening parameter. “Previously, scientists had to manually adjust this parameter and judge by eye what was the best sharpened or blurred map,” said Adams. The scientists describe their automatic measure of map quality in a recent publication (Acta Cryst. (2018) D74, 545-559). This tool, the phenix.auto_sharpen program, performs this process in a fully automatic way. Testing has shown that the program is more accurate than other available methods, and is less subjective than visual inspection, Adams noted.
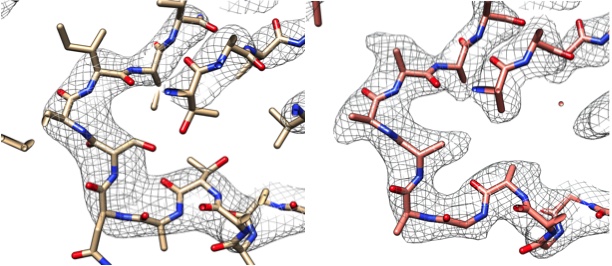
Left: Part of the cryo-EM map (gray grid) and model (protein chain) of the enzyme lactate dehydrogenase14 deposited in the EMDB and PDB (EMDB-8191, PDB 5K0Z). Right: Using Phenix, the map from the left was sharpened automatically and the sharpened map is presented with the automatically generated model.
After the map is sharpened, an atomic model is built from the cryo-EM experimental map. The phenix.map_to_model program, which was introduced in the recent Nature Methods article, can build the protein and nucleic acid chains of a macromolecular complex from a map and sequence in a fully automatic manner.
Refinement is a crucial step in model determination. “Once the atomic model is built,” said Adams, “it needs to be refined against the cryo-EM map. The atomic coordinates and other properties need to be optimized to achieve the best possible match between model and experimental map.” However, all of the atomic characteristics of the amino acids and nucleic acids must be preserved. The Phenix program that addresses this step in the process is phenix.real_space_refine, described last year (Acta Cryst. (2018). D74, 531–544). “The great thing about this program is that it allows for direct refinement against a cryo-EM map, without repurposing X-ray crystallography tools,” Adams added.
“Part of the reason for developing Phenix, in addition to giving newer users the same chance to produce an accurate model as an expert user, is to improve the overall quality of 3D structures that are available,” Adams said. Phenix developers found that they could improve the majority of cryo-EM structures that have already been deposited in the Protein Data Bank and Electron Microscopy Data Bank using their refinement program in automatic mode.
Model validation is vital throughout all stages of the structure solution process. Map validation helps the user understand the resolution of the map and set expectations for how accurate the final structure is likely to be. Validation of how well the model fits the map ensures the atomic model interprets the experimental data feasibly. Said Adams, “It is this step, atomic model validation, that helps electron microscopists obtain a final model that is chemically and physically meaningful.”
This suite of automatic tools is available from the Phenix website.
Pavel Afonine, Dorothee Liebschner, Nigel Moriarty, Billy Poon, and Oleg Sobolev develop Phenix at Berkeley Lab under the direction of Paul Adams. The other group leads are Tom Terwilliger, LANL; Jane and Dave Richardson, Duke University; and Randy Read, Cambridge University. Phenix is funded by the National Institutes of Health General Medical Sciences and the Phenix Industrial Consortium.

