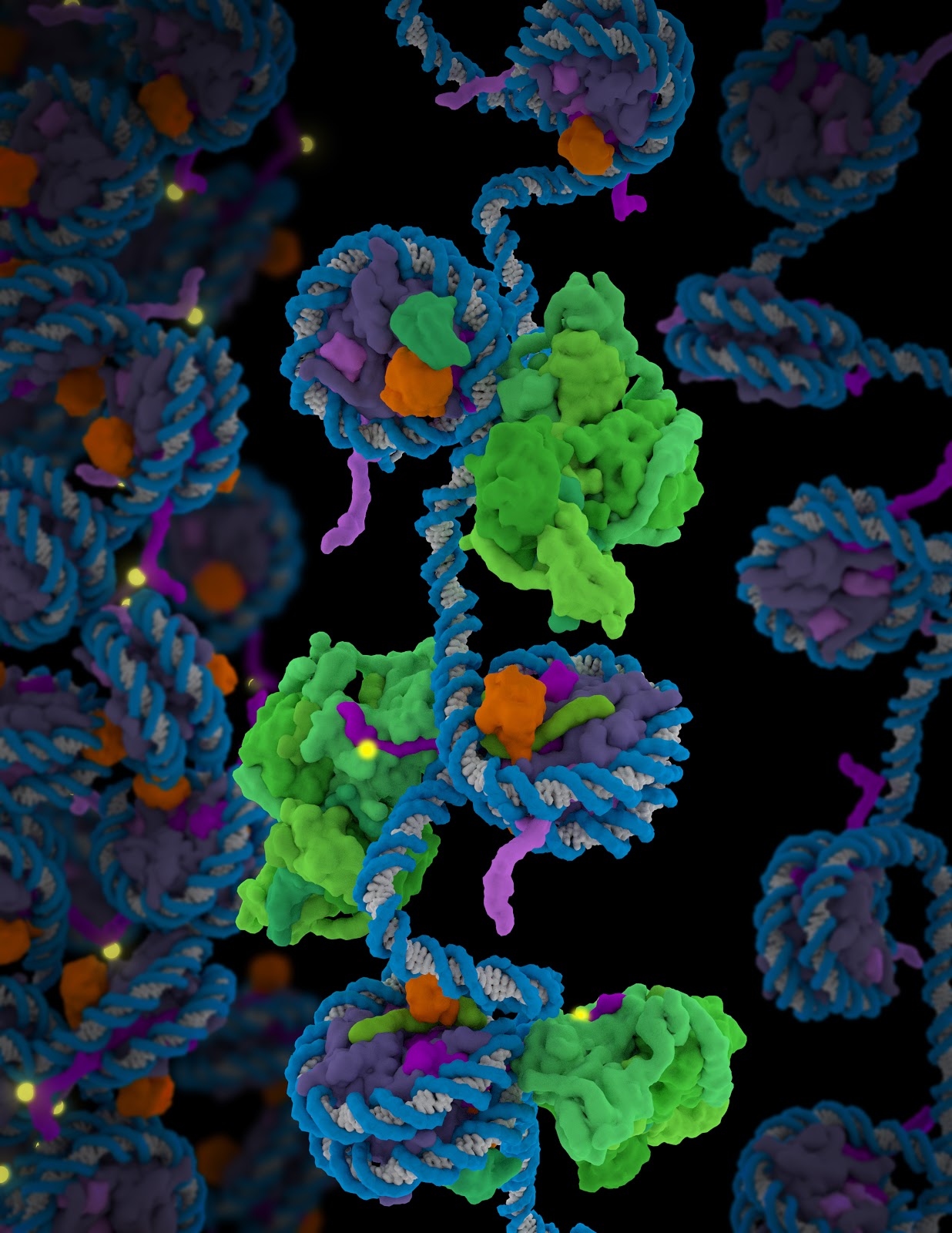
The genetic content within our chromosomes is highly organized. DNA is packaged into nucleosomes by wrapping of the double helical strands around proteins called histones. In turn, nucleosomes are packaged into higher-order chromatin structures that can be compacted or open—and thus determine if gene is silenced or active. Enzymes act to keep genes silenced by chemically marking the specific amino acids in histones with methyl groups that ultimately lead to chromatin condensation.
The multi-protein structure polycomb repressive complex 2 (PRC2) is involved in “silencing” genes so that they are not “read” by the cellular machinery that decodes genetic information, effectively keeping the genetic information in the “off” state. PRC2 silences genes by chemically depositing tri-methylation marks on histone H3 at lysine 27. Failure to regulate the activity of PRC2 not only impairs the process of development, but also contributes to the reversal of cell differentiation and the uncontrolled cell growth that are the hallmarks of cancer.
A team of scientists at Berkeley Lab and UC Berkeley have uncovered the molecular basis for the recruitment of PRC2 to certain locations of the genome and for the regulation of its activity. In a study published January 22 in Science, the researchers describe the structure of PRC2 while bound to a biologically relevant chromatin target. Using cryo-electron microscopy (cryo-EM), they uncovered crucial structural and functional information about this key regulator of cell differentiation and identity.
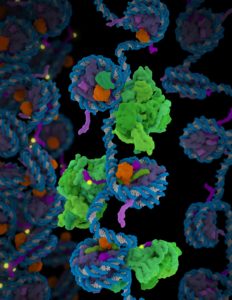
Molecular model showing polycomb repressive complex 2 (PRC2) in green interacting with nucleosomes: histone H3 tail shown in purple, methylation site in yellow and ubiquitin in orange. (Credit: Janet Iwasa, University of Utah)
The tail of histone H3, part of the chromatin in the structure elucidated by the researchers, is the region of the histone that is targeted by PRC2 for modification through an enzymatic process involving the transfer of a methyl group. “In the structure, we were able to localize the positions of mutations frequently found in various cancers,” said Eva Nogales, senior faculty scientist in the Molecular Biophysics and Integrated Bioimaging (MBIB) Division. “These mutations were found in the region of the complex that engages not only the substrate of its methyltransferase activity, the tail of histone H3, but also in regions responsible for chromatin binding, underscoring its functional relevance.”
Previous biochemical studies had indicated that histone methylation was one of the chromatin marks important for stimulation of PRC2 enzymatic activity. Nogales, who is also a Howard Hughes Medical Institute investigator and professor of Molecular and Cellular Biology at UC Berkeley, and her team, including former postdoctoral fellow Vignesh Kasinath (currently an assistant professor at the University of Colorado, Boulder), had published previous studies on this complex showing how cofactors of PRC2 mimic histone methylation to stimulate PRC2 activity.
In contrast to the earlier study, this new structure of PRC2 includes nucleosome containing a ubiquitination mark of histone H2A, as well as the protein cofactors JARID2 and AEBP2. The team observed a completely new region in the complex, the “bridge helix,” which connects the active module of the complex (its methyltransferase center) to the nucleosomal DNA and the histone tail in the nucleosome that it modifies. “The information we got from PRC2 and its interactions with other components in the structure was amazing,” said Nogales.
Nogales and her team were also able to generate exquisite pictures of how the different cofactors mediated the interaction of PRC2 with nucleosome. “The observation that JARID2 and AEBP2 each independently interact with the ubiquitin molecule on the histone H2A suggests a direct role for both cofactors in recruiting PRC2 to specific regions of the genome” said Kasinath, lead author and co-corresponding author on this study. This has implications for understanding how different histone modifications and cofactors come together to not only facilitate PRC2 recruitment but also regulate its gene silencing activity.
“In addition to the biological insights that are gained by studying this complex, we can understand the relevance of certain protein modules in the functionality of the complex and put cancer mutation in this context, which can lead eventually to therapeutics.” said Nogales. The findings from these studies open up new possibilities for combating cancer, while simultaneously expanding our knowledge of gene regulation at a molecular level.
The entire team involved in this study was: Curtis Beck, Paul Sauer, Jennifer Kosmatka, and Daniel Toso of UC Berkeley; Simon Poepsel of the University of Cologne; and Marco Faini and Ruedi Aebersold of ETH Zürich. The work was partially supported by the National Institutes of Health.

