In recent years, cryo-electron microscopy (cryo-EM) technology has advanced to the point that it can produce structures with atomic-level resolution for many types of molecules. Yet in some situations, even the most sophisticated cryo-EM methods still generate maps with lower resolution and greater uncertainty than required to tease out the details of complex chemical reactions.
In a study published in Nature Methods, a multi-institutional team led by Tom Terwilliger from the New Mexico Consortium and including researchers from Berkeley Lab demonstrates how a new computer algorithm improves the quality of the 3D molecular structure maps generated with cryo-EM.
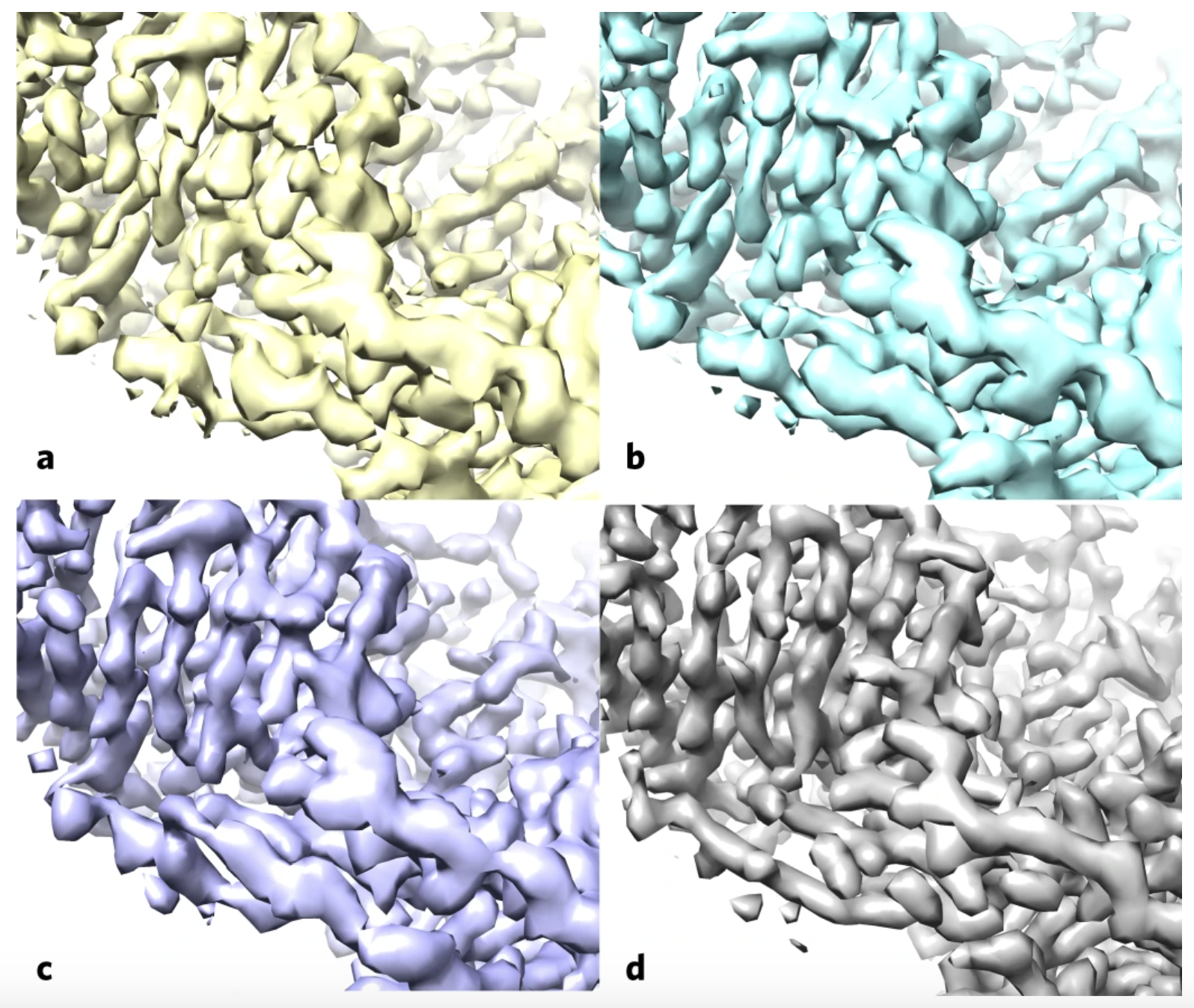
Using the enzyme β-galactosidase, also called lactase, as a test case, the researchers applied the standard methods (a) and then applied the improvement algorithm without (b) and with a filter to improve the uniformity of noise in the map (c), both of these maps are more similar to the deposited high-resolution protein structure map (d). (Credit: Terwilliger et al./Nature Methods)
The algorithm sharpens molecular maps by filtering the data based on existing knowledge of what molecules look like and how to best estimate and remove noise in microscopy data. An approach with the same theoretical basis was previously used to improve structure maps generated from X-ray crystallography, and scientists have proposed its use in cryo-EM before. But, according to study co-author Paul Adams, Director of the Molecular Biophysics and Integrated Bioimaging (MBIB) Division, no one had been able to show definitive evidence that it worked for cryo-EM until now.
The authors note that the clear benefits of the algorithm in revealing important details in the data, combined with its ease of use – it is an automated analysis that can be performed on a laptop processor – will likely make it part of a standard part of the cryo-EM workflow moving forward. In fact, Adams has already added the algorithm’s source code to the Phenix software suite, a popular package for automated macromolecular structure solution for which he leads the development team.
Read more in the Berkeley Lab News Center.

